I’ve been playing with the microelectronic example that came along with the geant4 package. I am using the original geometry from the microelectronics example, however, I shifted the example to the positive x-axis just to make my analysis process more convenient.
My Question
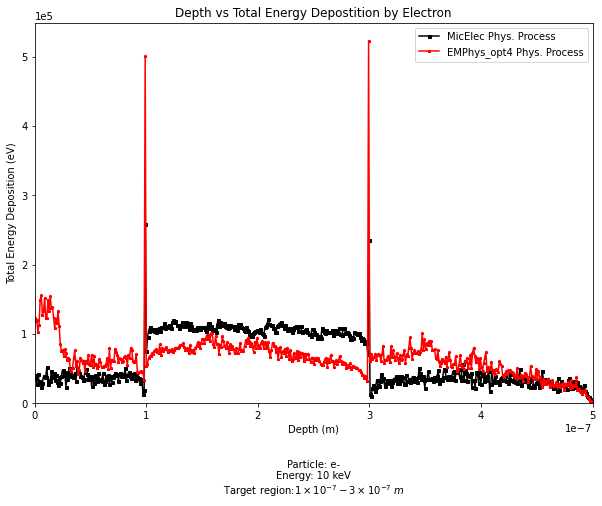
After firing 10,000 e- particles with energy 10 keV towards the target, I wrote the data into a CSV file and analyse them accordingly with python. Where I’ve noticed that at the boundary between the world and the logical target there seems to be something that is causing the energy of the electron deposited to be significantly larger than the other areas.
I then tried again with another physics list, and I seem to get the same result. (Please see the graph below)
Can anyone please point out what could be possibly causing this?
Are you recording the energy deposit at the post-step point? In reality, the “energy deposit” of a step really includes energy that was lost all the way along the step’s trajectory. Depending on your geometry, this could make a substantial difference in the energy/depth profile.
I am using the data I get from the GetTotalEnergyDeposit() method under the G4Step class to fill a CSV file. Could that be what is causing the problem?
G4Step::GetTotalEnergyDeposit() is correct. But I will bet that you are assigning that value to a single point, either the post-step point or the pre-step point. Right? In fact, that GetTotalEnergyDeposit() should be spread out along the whole line from the pre-step point to the post-step point.
I am not sure what exactly do you mean by spreading that out across the whole line from the pre-step point to the post-step point. It would be nice if you would like to point out the changes in an example. My current code goes something like this, (the line of interest is near the bottom)
There’s no great way to do this “properly” with changing your N-tuple structure and your analysis code. It’s not a coding issue, it’s a physics issue. You will need to come up with an approach that meets your own analysis needs.
Let me try to describe the conceptual problem a bit differently. Think about what the “actual particle” is doing as it traverses your volume. As it moves through the material, it is continuously losing energy, a little bit at a time (this is why we call it “dE/dx”, energy loss per unit length). Geant4 takes the particle’s trajectory and breaks it into steps. For each of those steps, Geant4 “integrates” dE/dx along the step, and gives you that total as “GetTotalEnergyDeposit()”. But that energy wasn’t all deposited at a point (your (x,y,z) coordinates in theN-tuple.
The problem as I understand it is that these steps as @mkelsey described are forcefully terminated at each volume boundary, hence the peaks at these positions!?
I know this is an old topic but I am running into a similar issue. My geometry is thin films with layers as little as 0.7 nm and up to 60 nm. I have looked at reducing the step size to force more frequent energy deposition, but I think that this method might work.
Do we think that it would be more accurate to deposit the energy at a single random point between pre-step and post step point, or do spread the energy out evenly over each bin between the pre and post step points?