Please fill out the following information to help in answering your question, and also see tips for posting code snippets. If you don’t provide this information it will take more time to help with your problem!
Geant4 Version: 11.3.1
Operating System: Ubuntu 22.04
Compiler/Version: GCC 11.4.0
CMake Version: 3.22.1
Hello,
I’m trying to use the molecularDNA example to compute DNA damage caused by monoenergetic C-12 ions. I only modified the source definition in the human_cell.mac macro (no other parts were changed). My source settings are:
/gps/particle ion
/gps/ion 6 12 6
/gps/pos/type Volume
/gps/pos/shape Ellipsoid
/gps/pos/centre 0 0 0 um
/gps/pos/halfx 9 um
/gps/pos/halfy 3.8 um
/gps/pos/halfz 9 um
/gps/ang/type iso
/gps/energy 100 MeV
/run/beamOn 20
/gps/source/list
In addition, I run the example with the following script:
rm -rf build
mkdir build
cd build
cp -r “/home/hezi/Downloads/geometries” .
cmake ..
make -j128
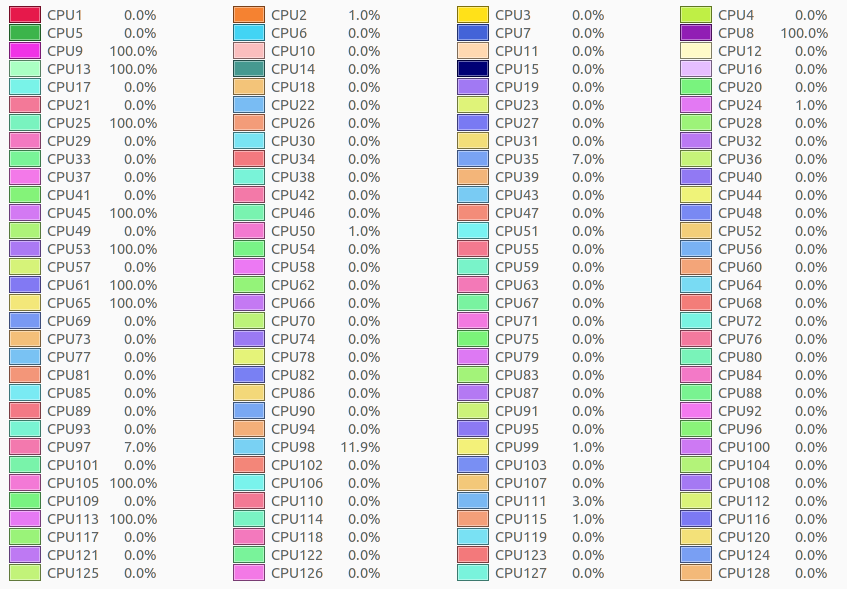
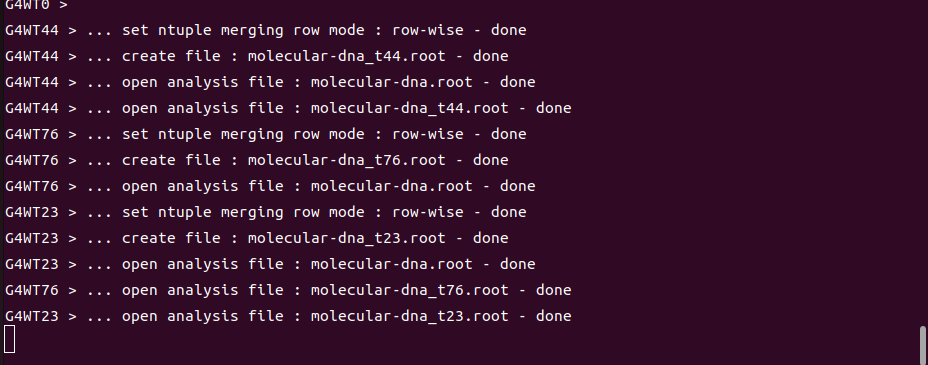
./molecular -m human_cell.mac -t 128 -p 6
hadd -O -f molecular-dna.root molecular-dna_t\*.root
root
.X human_cell.C
When I execute the program with this script, it has been running for over 72 hours and I still do not get any DNA damage data or histograms. I would like to understand what might be wrong.
Hardware configuration:
CPU: AMD® Epyc 7542 32-core processor × 128
RAM: 256 GB
Could you please advise what could cause the lack of outputs in this setup, or if additional configuration is required to use C-12 ions with the molecularDNA example?
Thank you!